Computer-Aided Drug Design (CADD)
As a modern computational technique, CADD technology comprises a broad range of theoretical approaches and in silico methodologies including molecular modeling and design, quantitative structure-activity relationships studies and so on. CADD enables to minimize the synthetic and biological experiments efforts by using modelling techniques and data analysis. In CADD, scientists use designed algorithms to calculate and analyze the relationship between drug molecules and receptor macromolecule to identify and develop a potential lead compound.
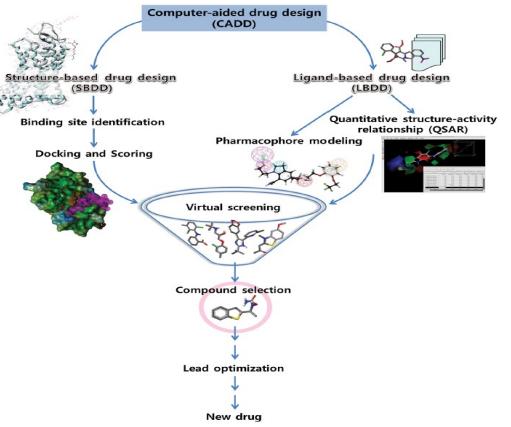 Fig.1 Computer Aided Drug Design. (Prajapat, P.; et al. 2020)
Fig.1 Computer Aided Drug Design. (Prajapat, P.; et al. 2020)Advantages of CADD
Compared to HTS, CADD can deliver multiple promising candidates requiring minimal compound.
CADD guides the development of huge virtual compound collections and thousands of compounds are allowed be screened at once in a short time.
A large number of drug candidates with undesired bioactivity can be filtered by CADD before entering experimental testing, which helps to save research time.
CADD provides comprehensive approaches to determine the structure of the receptor and pharmacophore of the ligand, giving rational guidance on the lead design.
A diversity of pharmaceutical properties of a potential drug are investigated, supporting the optimization process.
Once a functional molecule is determined, it can be grown into a new chemotypes by using CADD and a novel compound can be quickly designed in silico.
Development time and costs can be reduced significantly since there's no need of animal and human models.
Our Services of CADD
Our computational chemistry team is proficient in diverse disciplines including artificial intelligence, theoretical calculations, cloud computing and biochemistry. BOC Sciences is committed to offer a broad range of CADD services.
Quantum mechanics (QM)
Quantum mechanics is applied to determine the structure of proteins and study molecular mechanics. Scientists use this powerful tool to understand the reactions involving proteins with high accuracy and perform the ligand optimization. At BOC Sciences, we provide different and high quality QM calculation services to comprehensively describe the protein system including protein-ligand interactions and structures.
Structure-based virtual screening (SBVS)
SBVS is an important computational approach widely used in searching for new bioactive molecules against a certain target protein. It covers molecule collections screening, modeling and other computational methodologies. Our teams perform the SBVS by quickly conducting a large-scale searching of compound libraries, docking procedure and finally evaluating their binding affinity.
Ligand-based virtual screening (LBVS)
Unlike SBVS, LBVS dose not use the structural information of a biological target and it conducts the hit searching based on the information of the known ligand. In LBVS, we conduct searching of compounds that are ranked on the basis of similarity to the ligand and deliver compounds exhibiting similar biological properties.
De novo drug design
De novo drug design is an efficient method for the discovery of a variety of different biological target classes. It aims to generate new chemical molecules with desired properties in a more time-saving and cost-effective way. At BOC Sciences, we have established both structure-based and ligand-based approaches to accelerate drug discovery process and we are also equipped with a diversity of advanced computational tools and mathematical software.
ADME/Tox prediction
Absorption, distribution, metabolism and excretion (ADME) properties and various toxicities (T) cover many pharmacokinetic issues that determine whether a drug candidate can interact to target protein and have a effect in human's bodies. Prediction and evaluation of the ADMET are carried out to accelerate the research process and increase the success rate. We perform various assays to investigate parameters including membrane permeability, metabolic stability and toxicity, and we also conduct a rapid and accurate prediction from the structures of the molecule.
CADD Tools at BOC Sciences
Software
CHARMM, AMBER, NAMD, GROMACS and OpenMM (MD Simulation).
Auto Dock, GOLD, Cdocker, Mol Dock, Ligand Fit, PLANTS, Molegro Virtual, Docker and ICM (random searching).
FRED, DOCK, GLIDE, EUDOC, FLOG, SLIDE, ADAM, FlexX and eHiTS (systematic searching).
MODELLER, SWISS-MODEL (homology modeling).
Databases
Virtual compound collections containing millions of compounds.
Our Advantages of CADD
We are equipped with most advanced artificial intelligence and cloud computing to support your drug discovery projects.
Our integrated CADD platforms have delivered a great number of medicinal projects with high success rate and relatively low research cost.
Reference
Prajapat, P.; et al.Significance of Computer Aided Drug Design and 3D QSAR in Modern Drug Discovery. Journal of Medicinal and Organic Chemistry. 2017, 1.
※ It should be noted that our service is only used for research.

One-stop
Drug Discovery Services
- Experienced and qualified scientists functioning as project managers or study director
- Independent quality unit assuring regulatory compliance
- Methods validated per ICH GLP/GMP guidelines
- Rigorous sample tracking and handling procedures to prevent mistakes
- Controlled laboratory environment to prevent a whole new level of success
Online Inquiry