Quantum Mechanics for Target Selection
QM is an important tool in computer-aided drug design research, which attempts to explain the properties of atoms and molecules and their fundamental particles like protons, neutrons, electrons, etc. The properties of these particles such as their interactions with each other offer crucial information on the structure and performance of molecules. Scientists therefore usually use multiple physico chemical parameters obtained by QM technique to give a comprehensive characterization of a chemical molecule, which plays an essential role in understanding its reactivity.
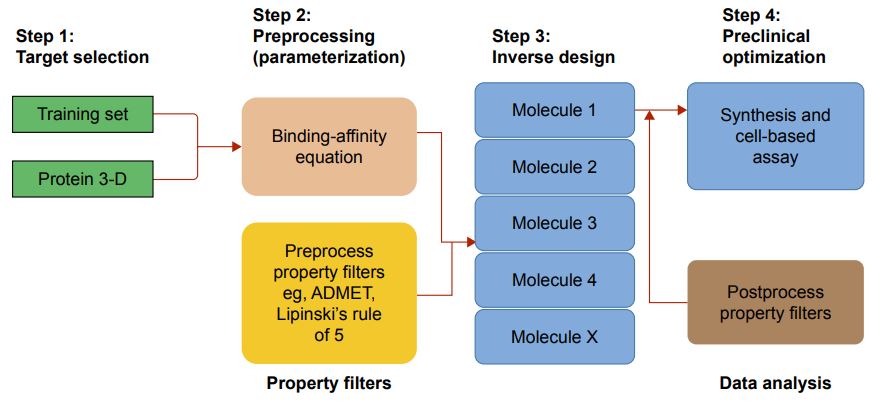 Fig.1 Implementation of QM in drug design workflow. (Olayide, A. A.; Mahmoud, E. S. 2017)
Fig.1 Implementation of QM in drug design workflow. (Olayide, A. A.; Mahmoud, E. S. 2017)Advantages of Quantum Mechanics
Compared to docking or quantitative structure-activity relationship method, QM owns the following merits:
QM can explore more binding mechanism details.
QM generates more accurate representation of molecular systems.
QM shortens the timeline of candidate compound discovery and optimization significantly.
Applications of Quantum Mechanics
To help determine the structure and performance of stable or unstable molecules.
To learn the relationship between structure and performance.
To investigate the interaction between molecules such as the collision.
Our Methods of Quantum Mechanics
QM/MM calculations
Our chemists have designed QM/MM calculations with high accuracy to study ligand binding.
Force field parameters from Ab initio calculations
We perform modeling studies on interactions between drug candidates with various biological targets including receptors, enzymes or other bio-macromolecules in large chemical systems.
We apply ball-and-spring models to describe molecules and the field forces can be calculated from potential energy functions such as bond lengths, etc.
Our Abilities of Quantum Mechanics
We are able to characterize the protein and solvent by representing the ligand and molecular mechanics with the knowledge of quantum chemistry.
Our teams are capable of conducting a diversity of calculations such as quantum chemical calculations, catalytic reaction mechanism calculation, transition state search and energy calculation, chemical reaction pathway and potential energy surface calculation, spectrum calculation, resonance frequency calculation, calculation of thermodynamic properties and enzyme catalytic mechanism simulation
We can elucidate reaction mechanism elucidation as well as provide optimization services.
Our data of QM method is also can be applied to support molecular design and virtual screening.
Our Advantages of Quantum Mechanics Services
Professional scientists at BOC Sciences have rich experience in conducting high accuracy QM/MM calculations and saving the modeling time.
We have designed multiple modern quantum molecular algorithms allowing for efficient searching of the chemical spaces.
Reference
Olayide, A. A.; Mahmoud, E. S.Quantum mechanics implementation in drug-design workflows: does it really help? Drug Design, Development and Therapy. 2017, 11: 2551-2564.
※ It should be noted that our service is only used for research.

One-stop
Drug Discovery Services
- Experienced and qualified scientists functioning as project managers or study director
- Independent quality unit assuring regulatory compliance
- Methods validated per ICH GLP/GMP guidelines
- Rigorous sample tracking and handling procedures to prevent mistakes
- Controlled laboratory environment to prevent a whole new level of success
Online Inquiry