Molecular Docking
Molecular docking simulation is an indispensable computational method in computer-assisted drug development. It aims to predict the binding mode of a compound with a given receptor of known structure, often achieved by assessing the candidate dockings through a scoring function.
Applications of Molecular Docking
- Help to understand the structural information of a protein complexed with its binding partner.
- Give a clear elucidation of the interaction mechanisms between proteins and other molecules (such as proteins, nucleic acids, small molecule compounds, peptides, etc.).
- Characterization of the binding events of small molecules and the target proteins.
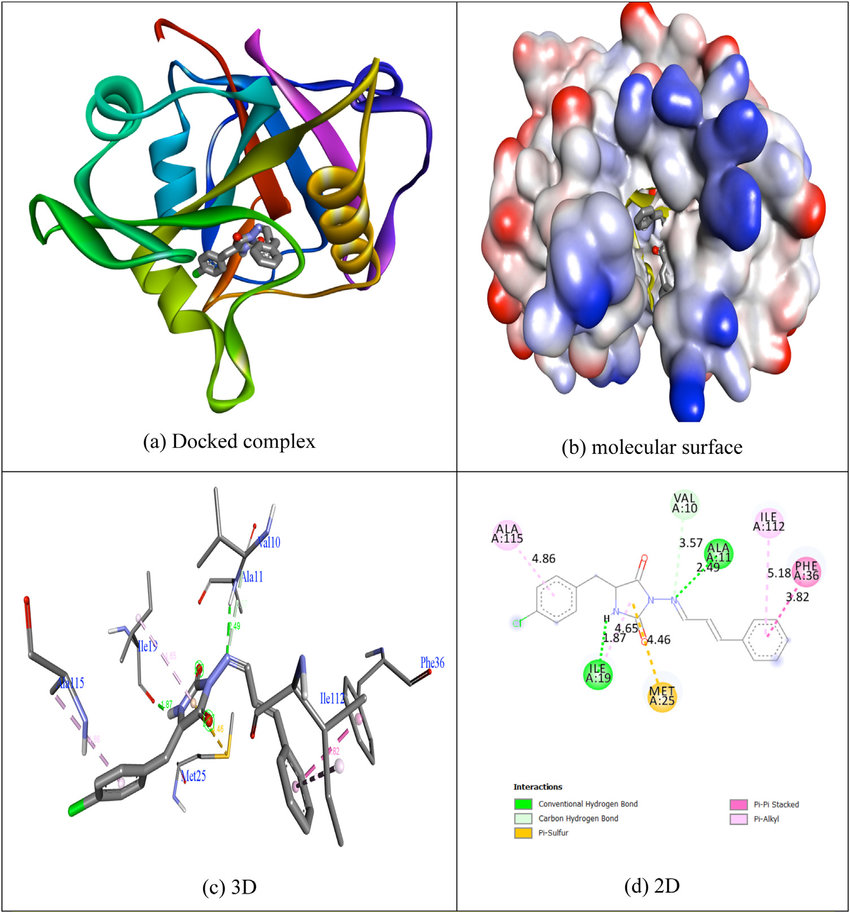 Fig.1 Molecular docking interaction of a compound within the binding site of the target protein. (Da, A.; et al. 2020)
Fig.1 Molecular docking interaction of a compound within the binding site of the target protein. (Da, A.; et al. 2020)Our Molecular Docking Services
At BOC Sciences, we have explored multiple computational docking techniques for various purposes to meet different demand of our customers.
Protein-ligand docking
We conduct protein-ligand docking for the detection and measurement of binding interactions between targets and their ligands, supporting the identification of drug leads by giving reliable prediction of behavior of protein molecules.
Protein-protein docking
Deep understanding of protein-protein interactions helps to find and identify disease-implicated targets. At BOC Sciences, we have developed fast fourier transform based algorithm to realize the protein-protein docking and conduct the searching and evaluation of all possible binding modes.
Protein-nucleic acid docking
Docking a small protein to nucleic acid receptors helps to investigate the interactions between them which is important in the identification of crucial amino acids or nucleotide residues. Our professional groups provide the docking services by predicting the complexes of 'protein-nucleic acid' structures consisting of docking, scoring and refinement.
Protein-peptide acid docking
Peptide docking can be regarded as a subproblem of protein-protein docking, in which the bound conformation of the peptide needs to be modeled. We present our well-developed peptide docking approach to provide you deeper insight into the protein-peptide interactions. Our teams offer targeted drug design and their structural characterization by predicting the complex structure accurately.
Antibody-antigen docking
Compared to structural determination technologies such as X-ray crystallography, computational methods offer a fast and reliable approach for generating structural models for such complexes. We provide high-quality antibody-antigen docking services to elucidate the structural basis of antibody-antigen interactions, supporting your rational design of efficient biological drugs based on antibodies.
Protein-lipid docking
Membrane proteins play critical roles in a diversity of cellular functions. Lipids, another major components of a membrane, have attached to target proteins tightly and interactions between these two molecules can provide may important functions. We introduce our well-developed docking technologies to investigate the complexity of lipid-protein interactions.
Protein-carbohydrate docking
As a key component of many biological processes, carbohydrate recognition is important in the targets discovery. The physiological and pathological significance of glycan-protein interactions play essential roles in the field of structure-based drug design. Protein-carbohydrate docking is one of essential techniques in computer-aided drug development. At BOC Sciences, we have established a novel and validated docking method which is specifically designed to handle docking of carbohydrate-like compounds.
Our Advantages of Molecular Docking Services
- BOC Sciences has equipped with high-performance computing equipment and advanced docking software to perform numerous calculations.
- Our experts take various factors into consideration in our docking sercices, such as flexibility of the protein, the interaction model mediated by water molecules and metal ions.
- We are capable of predicting multiple binding modes for proteins and different kinds of molecules.
- Our molecular docking procedures work seamlessly with upstream and downstream computational modeling protocols.
Reference
- Da, A.; et al.Antimicrobial activity of novel 5-benzylidene-3-(3-phenylallylideneamino)imidazolidine-2,4-dione derivatives causing clinical pathogens: Synthesis and molecular docking studies. Journal of Infection and Public Health. 2020, 12: 1951-1960.
※ It should be noted that our service is only used for research.

One-stop
Drug Discovery Services
- Experienced and qualified scientists functioning as project managers or study director
- Independent quality unit assuring regulatory compliance
- Methods validated per ICH GLP/GMP guidelines
- Rigorous sample tracking and handling procedures to prevent mistakes
- Controlled laboratory environment to prevent a whole new level of success
Online Inquiry