Molecular Dynamics Simulation
This computational method is used to simulate the behavior of a complex biological system. Combing the force field (which describes all the interatomic interactions) and Newtonian equations (which provides the information of position and speed of atoms over time), MD simulations enable to describe motions, interactions, and dynamics at the atomic level in a molecular system. They are also used in the determination and validation of structures obtained from x-ray crystallography or NMR experiments.
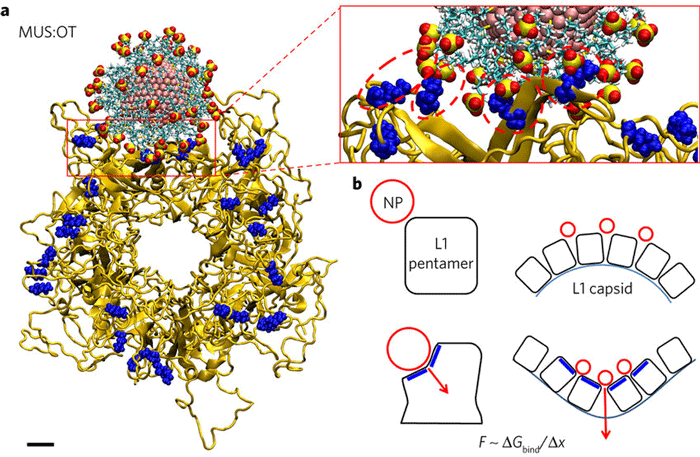 Fig.1 Molecular docking simulations. (Cagno, V.; et al. 2018)
Fig.1 Molecular docking simulations. (Cagno, V.; et al. 2018)The Application of Our MD Simulations Services
- We use the MD simulation methods to provide detailed information on the fluctuations and conformational changes of proteins and nucleic acids and study the protein system of your interesting.
- We can map ligand binding sites and analyze binding modes, providing a convincing strategy for regulating target activity through these ligands.
- We support the discovery of hidden binding sites and characterization of their druggability.
- Our experts help you have in-depth understanding of the affinity of the complex and quantify the energy changes in the whole process.
- We investigate the kinetics and thermodynamics of ligands binding to target proteins.
- At BOC Sciences, we can apply our MD simulations techniques in the generation of protein conformation sets.
- Assisting molecular docking
Before docking: We carry out MD simulations of protein targets and produce available protein conformational isomers different from the crystal structure.
After docking: We verify the binding pattern predicted by molecular docking and MD simulation is used as the final filter for hit optimization in the calculation and guiding chemical synthesis.
- Our MD simulation services are also able to assist the virtual screening, in which we introduce the protein flexibility before or after a docking protocol, refine the structure of protein-ligand complexes in the presence of water, ions, and even in membrane-like environments, and rank complexes with more accurate binding energy calculations.
Our Services of Molecular Dynamics Simulation
Material preparation
We select a model system in which missing segments are fixed and the protonation states determined.
We then equilibrate the system by using Newton's equations of motion until the system properties no longer change with time.
Calculation
After equilibration, we perform a production run to output trajectories and validate the properties of interest.
Optimization
We deliver a more accurate measurement by averaging the property over some time interval.
Our Capabilities of Molecular Dynamics Simulation
- Our MD simulations platform supports various force fields, such as CHARMM, AMBER, and OPLS.
- We can conduct the simulation of macromolecular systems which contain proteins, nucleic acids, lipids, etc.
- We are capable of performing high-performance program available for long time scale simulations
- At BOC Sciences, we offer visualization program for displaying, animating, and analyzing biomolecular systems.
- We carry out a conformational sampling and clustering of a target utilizing MD simulations before docking and explain the protein dynamics and the conformational selection by a ligand.
- In the simulation process, we validate the stability of the protein-ligand complex, identify persistent interatomic interactions and estimate the binding free energy.
- Our MD simulations include but not limited to steered molecular dynamics and interactive molecular dynamics.
- We apply the energy minimization based on steepest descent and conjugate gradient, and our calculation of free energy includes binding, solvation, interaction.
- We can apply our MD simulations in a more realistic model by including the induced-fit and solvent effects, which means it may apply even to a membrane-like environment in the case of membrane proteins.
Reference
- Cagno, V.; et al.Broad-spectrum non-toxic antiviral nanoparticles with a virucidal inhibition mechanism. Nature Materials. 2018, 17: 195-203.
※ It should be noted that our service is only used for research.

One-stop
Drug Discovery Services
- Experienced and qualified scientists functioning as project managers or study director
- Independent quality unit assuring regulatory compliance
- Methods validated per ICH GLP/GMP guidelines
- Rigorous sample tracking and handling procedures to prevent mistakes
- Controlled laboratory environment to prevent a whole new level of success
Online Inquiry