Machine Learning in Target Prediction
There's an urgent demand for a fast and effective method to distinguish drug-like and nondrug-like molecules from thousands of compounds obtained by the virtual screening. Therefore, machine learning techniques are rapidly developed for the purpose of classification and activity prediction in drug discovery studies. It is developed based on the high-dimensional data processing capability which is able to perform virtual screening of large compound libraries and classify molecules as active or inactive or rank these molecules based on their activity levels.
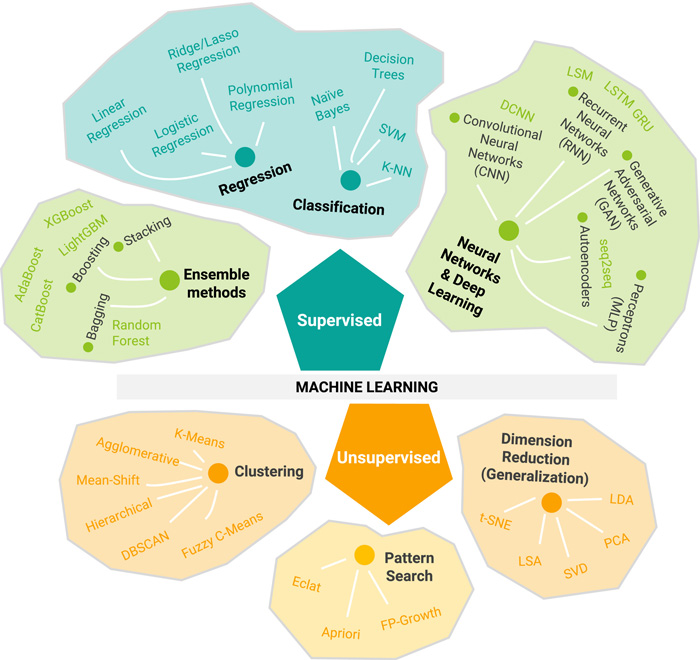 Fig.1 Overview of the types of machine learning and algorithms. (Talevi, A.; et al. 2020)
Fig.1 Overview of the types of machine learning and algorithms. (Talevi, A.; et al. 2020)Application of Machine Learning
Classification
Firstly, a machine-learning method takes a set of compounds that have previously been classified into two classes (in which these molecules are classified in to active ones or incative ones) as input.
Prediction
Then, these training-set molecules are analyzed and machine learning technique can develop a decision rule to classify new testing molecules into one of the two classes.
Our Machine Learning Services
We provide support vector machine (SVM) technique as well as binary kernel discrimination (BKD) method to generate new drug candidates.
Support vector machine
SVM technique has shown its strong abilities in the prediction of chemical and biological property. We have developed multiple SVM-based meta-classifiers and taken full advantages of their individual strengths, capable of performing computational identification of active compounds.
Binary kernel discrimination
BKD-based method is applied to conduct the virtual screening in databases of 2D structures that are represented by fragment bit-strings. It has robust function of compound selection using varying quantities of training data. Our experts can provide an effective and efficient BKD approach to perform the biological screening in your lead-discovery programs.
Our Advantages of Machine Learning Services
- We focus on the application of this supervised technique in prioritization of databases of molecules as active against a particular protein target.
- Our complex and accurate statistical learning algorithms enable to support the ligand-based similarity searching and structure-based docking.
- We also offer a set of services of data preparation, validation, optimization, and search methodologies to ensure its effectiveness.
Reference
- Talevi, A.; et al. Machine Learning in Drug Discovery and Development Part 1: A Primer. CPT: Pharmacometrics and Systems Pharmacology. 2020, 9(3).
※ It should be noted that our service is only used for research.

One-stop
Drug Discovery Services
- Experienced and qualified scientists functioning as project managers or study director
- Independent quality unit assuring regulatory compliance
- Methods validated per ICH GLP/GMP guidelines
- Rigorous sample tracking and handling procedures to prevent mistakes
- Controlled laboratory environment to prevent a whole new level of success
Online Inquiry