Protein Ligand Docking
Understanding how protein receptors recognize, interact and associate with molecular substrates and inhibitors is of vital importance in drug development. Protein-ligand docking aims to predict and rank the structure arising from the association between a given ligand and a target protein of known 3D structure. The process of docking a ligand to a binding site simulates the natural interaction between them via the lowest energy pathway. The goal is to find the best binding way which includes the binding sites and modes. Scientists use this modern method to predict the binding position when a ligand is bound to a to a protein receptor. Search algorithms and multiple scoring functions are the most popular docking tools in protein-ligand docking.
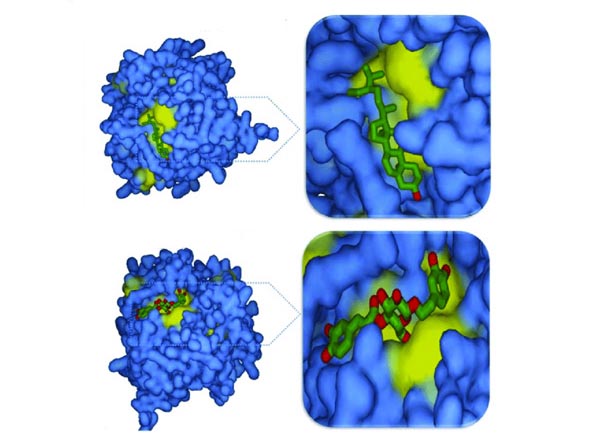 Fig.1 A picture of protein-ligand docking conformation.
Fig.1 A picture of protein-ligand docking conformation.(Mishra, R. K.; et al. 2016)
Our Docking Process And Services
System preparation
We prepare receptor with a known structure and ligand for the following docking process.
Docking
We apply complex search algorithm to generate a large set of small molecule binding to the active site. In this step, the identification of the active binding sites, prediction of the conformation and binding energy are able to achieved.
Docking calculation and scoring
We calculate the score of binding affinities which profiles the thermodynamics of interaction of the protein-ligand system, and rank them to obtain optimal structure.
Our Capabilities and Advantages of Protein Ligand Docking
Our computational chemistry teams can conduct fast sampling campaigns with efficient computing search algorithm parallel computing with efficient search.
We apply multiple scoring function including van der Waals, electrostatics, hydrophobic contacts, hydrogen bonds, surface complementarity, solvent effects, etc.
We are capable of performing various docking modes such as induced fit docking with partial flexibility of the receptor and multi-ligand docking with cofactor, water, metal ions.
Reference
Mishra, R. K.; et al. Exploration of anti-Malassezia potential of Nyctanthes arbor-tristis L. and their application to combat the infection caused by Mala s1 a novel allergen. Bmc Complementary & Alternative Medicine. 2016, 16(1): 114.
※ It should be noted that our service is only used for research.

One-stop
Drug Discovery Services
- Experienced and qualified scientists functioning as project managers or study director
- Independent quality unit assuring regulatory compliance
- Methods validated per ICH GLP/GMP guidelines
- Rigorous sample tracking and handling procedures to prevent mistakes
- Controlled laboratory environment to prevent a whole new level of success
Online Inquiry